集產品研發、生產、銷售于一體
多年產品研發經驗,自主知識產權
13671881166
400-6885-928

集產品研發、生產、銷售于一體
多年產品研發經驗,自主知識產權
13671881166
400-6885-928

第一作者:張廣吉
通訊作者:王立強副教授,劉又年教授
通訊單位:中南大學化學化工學院,鄭州大學材料科學與工程學院
論文DOI:10.1021/acscatal.2c01113
全文速覽
開發可替代貴金屬基催化劑、實現硝基化合物的高效加氫還原制備相應氨基化合物的非貴金屬催化體系具有重要意義但仍充滿挑戰。該工作報道了一種多級多孔碳載N,S共配位穩定的鈷單原子催化劑(Co1/NSC-AT),以實現硝基化合物高效加氫還原。Co1中心獨特的配位環境結合碳載體的多級孔結構,賦予了Co1/NSC-AT優異的催化性能;加氫反應可以在溫和的條件(35 oC, ~1 bar H2)下高效進行。理論計算表明,Co1/NSC-AT中的N,S共配位的鈷單原子中心(Co1S1N3)是氫化反應的活性中心。與Co1N4和Co納米顆粒相比,Co1S1N3具有較低的反應勢壘。此外,N、S共配位環境可以調整Co單原子的電子結構,促進H從活性位點上的解吸,從而促進加氫過程。
背景介紹
硝基化合物還原是制備氨基化合物及其衍生物的重要手段。以貴金屬為催化劑、氫氣為還原劑的催化加氫技術是當前硝基還原的使用最為廣泛的手段。然而,貴金屬基催化劑存在成本高、加氫選擇性差且易中毒等問題,限制了其大規模的推廣使用。因此,發展高效非貴金屬加氫催化劑是當前研究的趨勢。雜原子摻雜碳負載的Fe 系(Fe、Co 和 Ni)金屬,尤其是金屬中心具有原子分散特征的碳負載鐵系單原子催化劑,因其極低的成本、高選擇性和良好的催化活性,是最有前景的催化劑之一。目前,鐵系金屬催化劑面臨的挑戰是如何將碳負載單原子催化劑的活性提高至貴金屬基催化劑所具備的水平。該方面的研究雖然取得了一些研究進展,但單原子催化劑在硝基化合物的加氫反應中仍處于起步階段,其催化性能仍有極大的提升空間。
研究出發點
單原子催化劑的活性受其金屬中心的電子結構和空間分布影響。金屬中心的電子結構主要受配位原子影響,其對反應物的吸附/解吸、中間體的形成以及產物的解離都具有重要的影響。然而,當前報道的碳負載單原子催化劑的金屬中心多由N原子穩定。N原子電負性高,導致電子金屬中心呈現缺電子狀態,這對于反應物或中間體的吸附/解離并非總是優選的。另一方,并非所有單原子位點都是有催化活性的。單原子位點通常位于微孔結構中,僅有暴露在催化劑“外表面”的位點才是活性的。就此而言,介孔結構可以通過聯通微孔,而使活性充分暴露。為此,作者提出了一種利用蛋白-金屬離子網絡化學策略,構建多級孔碳負載的、N, S共配位鈷單原子催化劑,即Co1/NSC-AT。其兼具高效的反應位點Co1S1N3和多級孔結構帶來的高的活性位點利用率。
圖文解析
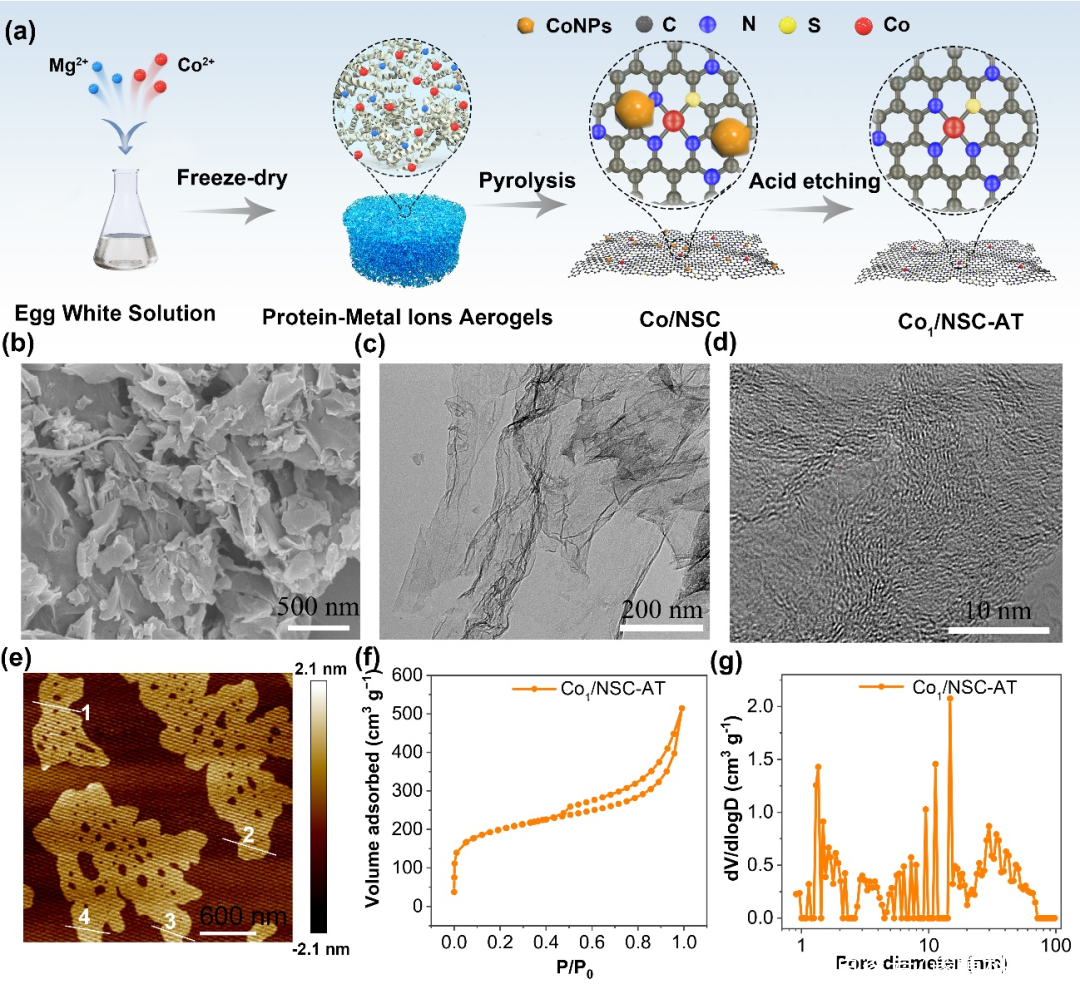
圖1. 催化劑Co1/NSC-AT的(a)制備示意圖.(b–e)掃描電鏡、透射電鏡、HR-透射電鏡和原子力顯微鏡圖像.(f) N2吸附等溫線和(g)孔徑分布.
Co1/NSC-AT的SEM,TEM和高分辨TEM分別如圖所示。Co1/NSC-AT呈現出幾何片狀結構,由彎曲的多孔超薄石墨烯構成,AFM測試結果證明樣品厚度有1.5~2.0 nm ,呈單層或少層石墨烯樣。Co1/NSC-AT顯示出多級孔結構,高的孔體積,大的BET比表面積。
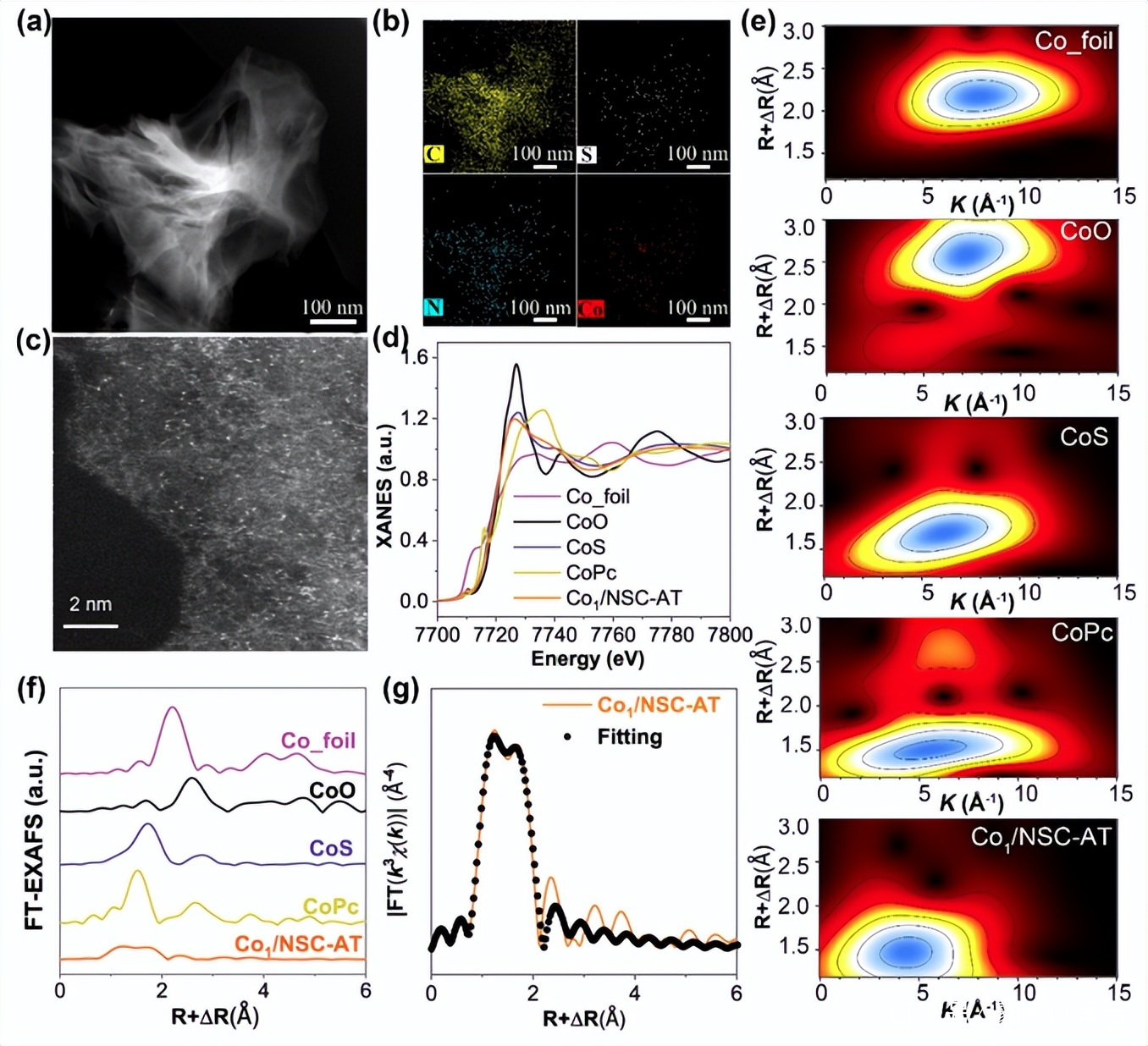
圖2. Co1/NSC-AT的(a)HAADF-STEM圖像和 (b) C、S、N 和 Co 的相應 EDX 元素映射 。(c)相差校正HAADF-STEM 圖像。(d) K 邊緣的 XANES 光譜。(e) 小波變換。(f) FT R空間曲線。(g)EXAFS的R空間擬合曲線。
Co1/NSC-AT的HAADF-STEM和相應的EDX mapping圖像顯示,Co,C,N和S均一的分布在碳基質中,而且金屬鈷以單原子形式存在,未發現鈷納米粒子。為了進一步探索Co原子的化學價態和配位環境,對Co1/NSC-AT進行了X射線吸收近邊結構(XANES)和擴展X射線吸收精細結構光譜測量。結果表明Co1/NSC-AT中的Co帶有正電荷,價態為~ +2。Co的 k邊傅里葉變換k3加權EXAFS光譜和EXAFS小波轉換分析可知,Co是單原子分散的,且被N、 S共配位穩定。EXAFS擬合計算出Co原子的配位數為4,N、S原子的比例為3:1,因此Co中心的理論配位結構為Co1S1N3,是催化劑Co1/NSC–AT中的主要催化活性位點。
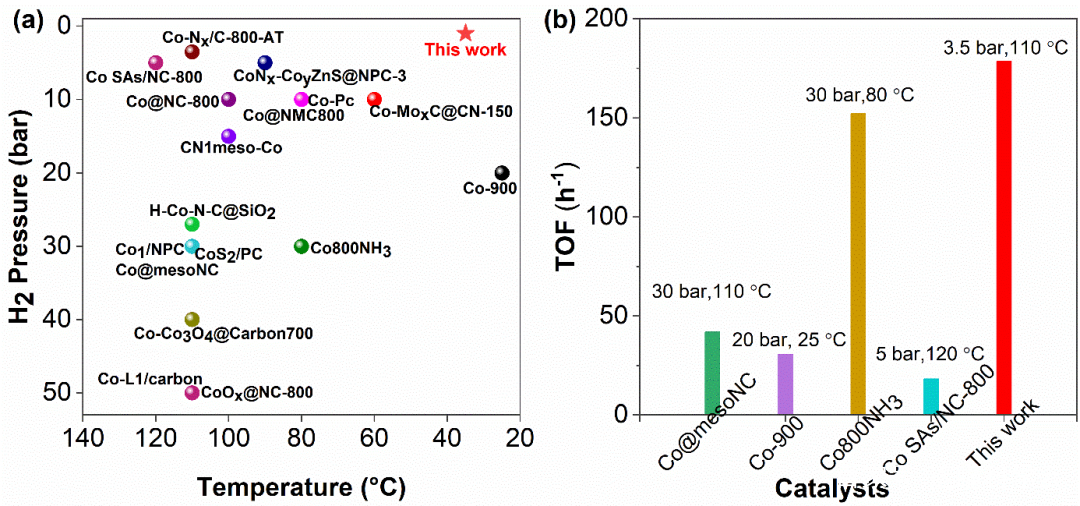
圖3 催化劑Co1/NSC-AT與近來文獻報道的鈷催化劑硝基加氫性能對比
為了考察催化劑Co1/NSC-AT對不同硝基底物的催化選擇性和普適性,進行了底物擴展實驗(45個底物)。大部分的硝基加氫反應都可以在溫和的條件下 (85 oC,5 bar)進行,轉化率達到99%,選擇性大于99%,優于最近報道的N配位穩定的鈷單原子催化劑。
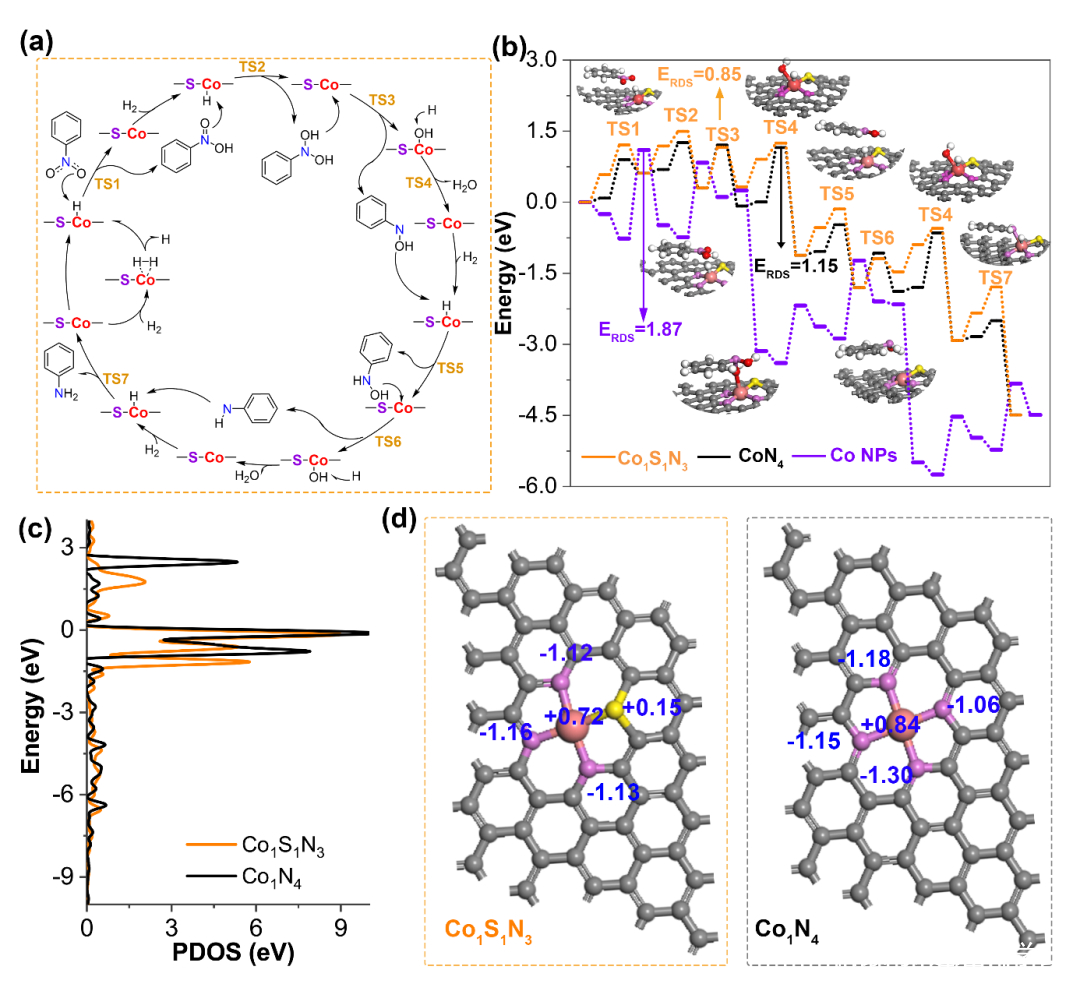
圖4. (a) 硝基苯 (PhNO2) 在Co1S1N3 位點加氫生成苯胺 (PhNH2)的反應途徑。(b) 在不同位點Co1S1N3,Co NPs和 Co1N4上硝基苯加氫的能量分布;(c) CoN4 和 Co1S1N3模型的DFT 計算預測狀態密度;(d) 不同Co中心的Bader電荷.
為了研究硝基化合物在單原子Co1S1N3位點的催化機理,對硝基苯的催化加氫進行了DFT 計算模擬。在Co1S1N3位點上對硝基苯的加氫催化遵循直接途徑,從C6H5NO2H2 到 C6H5NOH這一步在整個加氫反應過程中擁有最高的能壘 (0.85 eV),是反應的決速步驟。隨后,通過局部態密度 (PDOS) 和Bader電荷分析,揭示了配位環境對與其催化性能密切相關的電子結構的影響。在Co1S1N3中Co 3d軌道的PDOS與Co1N4 中的相比能量水平低,表明Co1S1N3與H*的結合能力較弱,有利于H的脫附,促進加氫反應。由于Bader電荷分析可知, Co1N4 和 Co1S1N3位點上Co原子的電荷密度分別為 +0.72 和 +0.84 e。這表明S的引入可以降低Co 的氧化態,導致其與 H 的相互作用較弱。總體而言,N、S 配位結構為單原子中心提供了獨特的電子結構,有利于硝基苯加氫反應的進行。
總結與展望
利用蛋白-金屬離子網絡構筑了多級孔碳負載的,N、S共配位穩定Co單原子催化劑,實現了硝基化合物的高活性、高選擇性加氫。研究結果將為碳負載單原子催化劑的結構設計及性能優化、高性能硝基加氫催化劑的構建提供思路。
通訊作者介紹
劉又年,中南大學化學化工學院教授,博士生導師。獲得省部級自然科學獎、科技進步獎等5項。多年來主要從事多功能納米材料的制備及其在生物醫用、藥物控釋、環境能源和光電熱催化中的應用。在Adv. Mater., Angew. Chem. Int. Ed., PNAS, Adv. Func. Mater., ACS Nano, AIChE J等刊物發表學術論文200余篇,他引7000余次,H-Index 42,授權國家發明專利20余項。
王立強,鄭州大學材料科學與工程學院副教授,碩士生導師。主要從事催化劑設計、鋁資源的高效利用等方面的研究。在ACS Catal. Adv. Mater., Angew. Chem. Int. Ed., ACS Nano, 等刊物發表學術論文40余篇,他引2000余次。擔任Journal of Central South University、Frontiers in Chemistry等雜志的青年編委或客座編委。
文獻來源
Guangji Zhang, Feiying Tang, Xin Wang, Liqiang Wang, and You-NianLiu. Atomically Dispersed Co–S–N Active Sites Anchored on Hierarchically Porous Carbon for Efficient Catalytic Hydrogenation of Nitro Compounds. ACS Catalysis,2022, 12, 5786–5794.
原文鏈接:
https://pubs.acs.org/doi/full/10.1021/acscatal.2c01113.
瀏覽次數:907
瀏覽次數:945
瀏覽次數:1161
瀏覽次數:1152
瀏覽次數:2320
